Уполномоченный Представитель изготовителя в Европейском Союзе

Мы принимаем документацию для оценки соответствия на латышском, английском. немецком, украинском, русском, литовском, румынском, венгерском языках.
Нам доверяют функции Уполномоченного представителя производителя многие компании, в том числе глобального уровня из США, Канады, Израиля, Австралии, Японии, Аргентины, Перу, России, Украины, Индии и Китая.
Выбор правильного уполномоченного представителя в Европе важен для поддержки устойчивого роста бизнеса.
European Authorized Representative (Authorised) является связующим звеном между вашей компанией и соответствующими национальными компетентными органами, чтобы обеспечить соблюдение требований.
Уполномоченный представитель ЕС будет действовать в качестве вашего основного контактного лица для всех компетентных органов.
Уполномоченный представитель ЕС хранит актуальную копию вашего Технического файла, доступную для проверки европейскими компетентными органами.
Наименование, адрес и контактные данные назначенного представителя будут указаны на упаковке и сопутствующих материалах продукта.
Информирование вашей компании о любых изменениях в соответствующих законах, которые повлияют на конкурентоспособность вашего продукта.
Если государство-член попытается вывести ваше устройство с рынка, уполномоченный представитель ЕС будет представлять вашу компанию в Европейской комиссии.
Уведомляет или получает уведомление от компетентных органов о серьезных инцидентах с продукцией.
По требованию производителя приложит все усилия, чтобы закупить продукт, который, возможно, стал причиной жалобы покупателя, и отправит упомянутый продукт для оценки соответствия.
Подписывает ЕС Декларации о соответствии под полную свою ответственность, если поручено производителем.
Отвечает за устранение несоответствий и корректирующих мерах безопасности на местах.
Помощь с технической документацией, включая анализ рисков.
Уполномоченный представитель ЕС также будет осуществлять любой необходимый послепродажный надзор, если это определено в договоре с производителем.
Есть еще много других обязанностей Уполномоченного представителя ЕС, которые необходимо выполнить, если определено контрактом, в том числе контакты с европейскими лабораториями и нотифицированными органами.
Компетентный надзорный орган может в любой момент проверить
European Authorized Representative посредством инспекций регулирующих органов, чтобы определить, понимают ли они свою роль, имеют ли они прямой доступ к документации клиента, к Техническому файлу и Проектному досье, и подготовили процессы, чтобы убедиться, что он может подтвердить соответствие требованиям продукции.
В контракте на обслуживание, который заключается с производителем, упоминается требование о страховании ответственности, это решение должно быть взаимно согласовано.
В любом случае, наша компания страхует риски от возможных ошибок, которые могут произойти в процессе работы.
Наши компетентные сотрудники оценят ваш технический файл и ответят на любые запросы или опасения компетентных органов. Мы регулярно предоставляем всем нашим клиентам из официальных представительств регулярные и исчерпывающие обновления о важных нормативных обновлениях для европейского рынка.
Наши персонал по вопросам европейского регулирования работают с компаниями по всему миру и представляют их в ЕС в качестве своих уполномоченных представителей. Мы можем представить вашу компанию и ваше устройство на европейском рынке.
Мы в состоянии предложить квалифицированных специалистов, обладающих необходимыми экспертными знаниями о нормативных требованиях ЕС практически во всех сферах. Знания наших специалистов подтверждаются академической квалификацией и профессиональным опытом в сфере нормативного регулирования.
Обладая фундаментальными знаниями европейских стандартов и опытом оценки соответствия, мы поможем вам упростить и оптимизировать процедуру оценки соответствия для рынка Европейского союза с правом нанесения на продукцию СЕ маркировки.
Наши эксперты дадут практические советы и предоставят поддержку от концепции до прототипа и далее, чтобы помочь вам вовремя вывести свою продукцию на рынок.
Мы говорим с вами на одном языке, что важно для понимания заказчиком исполнителя и наоборот и принимаем документацию на русском языке.
Наши эксперты в состоянии предоставить
• Бесплатную первичную консультацию для оценки требований и планирования оценки соответствия вашего продукта.
• Определение законодательных актов, основных требований безопасности и установление стандартов, которые применяются к продукту.
• Выполним анализ и оценим рисков по каждому пункту в соответствии с установленными стандартами.
• В случае отсутствия обоснования безопасности, составим и предоставим данный документ.
• Составим отчет о соответствии продукта применимым требованиям с указанием областей несоответствия и советами о том, как устранить любые обнаруженные пробелы, особенно связанные с электромагнитной совместимостью и электробезопасностью.
• Оценим соответствие технической документации требованиям Европейского союза.
• При необходимости предоставим информацию о процедурах соответствия.
• По вашему требованию проведем испытания продукции в аккредитованной европейской лаборатории.
• Проведем аудит производства.
• Составим отчет о соответствии продукта применимым требованиям с указанием областей несоответствия и советами о том, как устранить любые обнаруженные пробелы, особенно связанные с электромагнитной совместимостью и электробезопасностью.
• Предоставим проект декларации соответствия, если подписывает ее производитель.
• Выступим в качестве вашего уполномоченного представителя в ЕС, в том числе с подписанием под свою полную ответственность EU Declaration of conformity.
• Обновление технической документации и ЕС Декларации соответствия при изменении гармонизированных стандартов, Директив и Регламентов, даже после того, как продукт выведен на европейский рынок.
Свяжитесь с нами сегодня для первоначального разговора о представителе.
В случае, если наши специалисты не смогли ответить на вопросы, связанные с оценкой соответствия для рынка Европейского союза, можете обратиться в нотифицированный орган ЕС SIA International center for quality certification – ICQC или к экспертам Center for Testing and European Certification (CTEC).

Директивы Европейского союза
Европейский подход к оценке соответствия продукции
Уполномоченный представитель для медицинских изделий в Европейском Союзе
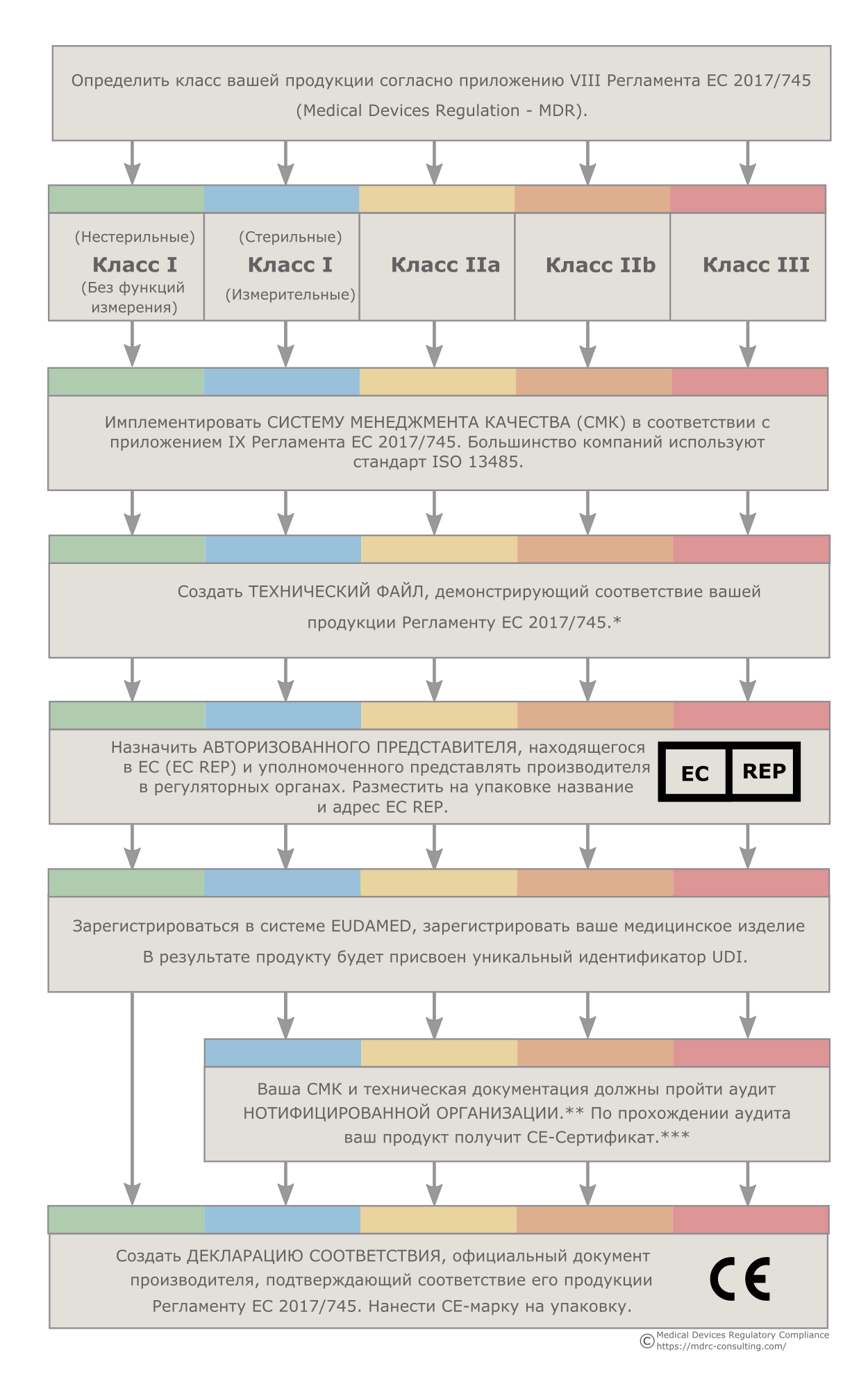
Согласно Европейскому Регламенту по медицинским изделиям (EU MDR 2017/745) и Регламенту по медицинским изделиям для диагностики in vitro (EU IVDR 2017/746), производитель, находящийся за пределами ЕС, прежде чем разместить свою продукцию на европейском рынке, должен назначить уполномоченного представителя в Европейском Союзе.
Вам требуется Уполномоченный представитель в ЕС?
Свяжитесь с нами:
Уполномоченного представителя в Европе также иногда называют EUAR, EC REP, CE REP, EU REP или EAR. Все эти названия используются специалистами по медицинскому оборудованию, но обозначают они одно и то же: Уполномоченный представитель ЕС.
Что означает Уполномоченный представитель в ЕС согласно MDR и IVDR
Уполномоченный представитель в Европе определяется как организация в Европейском союзе, которая приняла письменный мандат от не европейского производителя и согласилась действовать от его имени в отношении определенных задач в соответствии с MDR или IVDR. В частности, Уполномоченные представители играют ключевую роль в обеспечении соответствия устройств требованиям и служат контактным лицом для европейских властей и нотифицированных органов.
MDR и IVDR описывают задачи, которые производитель может делегировать уполномоченному представителю, и условия, при которых это может происходить. Эти отношения должны быть четко оговорены в договоре (мандате) между Уполномоченным представителем и производителем. Уполномоченный представитель выполняет задачи, указанные в мандате. Копия мандата предоставляется надзорным органам ЕС.
Уполномоченный представитель несет ответственность за проблемы, связанные с медицинскими изделиями производителя, находящиеся на европейском рынке, наряду с самим производителем.
Мы готовы стать вашим Уполномоченным производителем в ЕС.
Свяжитесь с нами
Задачи Уполномоченного представителя
MDR и IVDR четко определяют суть роли Уполномоченного представителя и его обязанности, которые должны быть указаны в мандате:
- проверять наличие технической документации и Декларации соответствия;
- проверять выполнение производителем всех процедур в соответствии с MDR или IVDR;
- держать у себя копии технической документации, Декларации соответствия и всех необходимых сертификатов;
- обеспечивать регистрацию в Eudamed;
- при необходимости, взаимодействовать с надзорными органами ЕС;
- обеспечивать коммуникацию между надзорными органами и производителем;
- обеспечивать коммуникацию в отношении превентивных и корректирующих действий;
- информировать производителя о жалобах и инцидентах, связанных с его продукцией.
Требования к Уполномоченному представителю
Уполномоченный представитель должен иметь в своем распоряжении лицо, ответственное за соблюдение нормативных требований (также известному как PRRC), которое обладает достаточным опытом в отношении европейских нормативных требований к медицинским устройствам или устройствам in vitro. Требуемый опыт такого лица:
А) признанный в ЕС диплом в области права, медицины, фармации, инженерии или другой соответствующей научной дисциплины, и, по крайней мере, один год профессионального опыта в области регулирования или в системах управления качеством, связанных с медицинскими изделиями;
В) четыре года профессионального опыта в области европейской регуляторики или систем управления качеством, связанных с медицинскими изделиями.
Если вы ищете Уполномоченного представителя в ЕС, свяжитесь с нами:
Или воспользуйтесь формой для контактов ниже:
ФОРМА ДЛЯ КОНТАКТОВ
Уполномоченный представитель для медицинских изделий в Европейском Союзе
Согласно Европейскому Регламенту по медицинским изделиям (EU MDR 2017/745) и Регламенту по медицинским изделиям для диагностики in vitro (EU IVDR 2017/746), производитель, находящийся за пределами ЕС, прежде чем разместить свою продукцию на европейском рынке, должен назначить уполномоченного представителя в Европейском Союзе.
Уполномоченного представителя в Европе также иногда называют EUAR, EC REP, CE REP, EU REP или EAR. Все эти названия используются специалистами по медицинскому оборудованию, но обозначают они одно и то же: Уполномоченный представитель ЕС.

Вам требуется Уполномоченный представитель в ЕС?
Что означает Уполномоченный представитель в ЕС согласно MDR и IVDR
Уполномоченный представитель в Европе определяется как организация в Европейском союзе, которая приняла письменный мандат от не европейского производителя и согласилась действовать от его имени в отношении определенных задач в соответствии с MDR или IVDR. В частности, Уполномоченные представители играют ключевую роль в обеспечении соответствия устройств требованиям и служат контактным лицом для европейских властей и нотифицированных органов.
MDR и IVDR описывают задачи, которые производитель может делегировать уполномоченному представителю, и условия, при которых это может происходить. Эти отношения должны быть четко оговорены в договоре (мандате) между Уполномоченным представителем и производителем. Уполномоченный представитель выполняет задачи, указанные в мандате. Копия мандата предоставляется надзорным органам ЕС.
Уполномоченный представитель несет ответственность за проблемы, связанные с медицинскими изделиями производителя, находящиеся на европейском рынке, наряду с самим производителем.
Мы готовы стать вашим Уполномоченным производителем в ЕС.
Задачи Уполномоченного представителя
MDR и IVDR четко определяют суть роли Уполномоченного представителя и его обязанности, которые должны быть указаны в мандате:
- проверять наличие технической документации и Декларации соответствия;
- проверять выполнение производителем всех процедур в соответствии с MDR или IVDR;
- держать у себя копии технической документации, Декларации соответствия и всех необходимых сертификатов;
- обеспечивать регистрацию в Eudamed;
- при необходимости, взаимодействовать с надзорными органами ЕС;
- обеспечивать коммуникацию между надзорными органами и производителем;
- обеспечивать коммуникацию в отношении превентивных и корректирующих действий;
- информировать производителя о жалобах и инцидентах, связанных с его продукцией.
Требования к Уполномоченному представителю
Уполномоченный представитель должен иметь в своем распоряжении лицо, ответственное за соблюдение нормативных требований (также известному как PRRC), которое обладает достаточным опытом в отношении европейских нормативных требований к медицинским устройствам или устройствам in vitro. Требуемый опыт такого лица:
А) признанный в ЕС диплом в области права, медицины, фармации, инженерии или другой соответствующей научной дисциплины, и, по крайней мере, один год профессионального опыта в области регулирования или в системах управления качеством, связанных с медицинскими изделиями;
В) четыре года профессионального опыта в области европейской регуляторики или систем управления качеством, связанных с медицинскими изделиями.
Если вы ищете Уполномоченного представителя в ЕС, свяжитесь с нами:
Или воспользуйтесь формой для контактов внизу страницы
Ec rep что означает маркировка
Определение Директив ЕС, требованиям которых должна соответствовать продукция:
93/42/ЕЭС – для медицинских изделий (MDD) or 90/385/ЕЭС — для активных имплантируемых медицинских изделий (AIMDD).
Определение классификации продукции с помощью приложения IX Директиву для медицинских изделий (MDD):
Класс I (нестерильные, без фунции измерения),Класс I (стерильные, с фунцией измерения),Класс IIa,Класс IIb или Класс III/AIMD*.
* Активные имплантируемые медицинские изделия подлежат тем же нормативными требованиями как изделия Класса III
Для всех устройств, кроме Класса I (нестерильные, без функции измерения), осуществить Систему Менеджмента Качества (СМК),
в соответствии с Приложением II или Приложением V директивы MDD.
Для продуктов Класса III/AIMD, подготовить Дизайн Досье (Design Dossier*).
Для всех остальных устройств, подготовить ЕС Технический файл, который содержит подробную информацию подтверждающую соответствие медицинского изделия
директиве MDD 93/42/ЕЭС.
* Для устройств Класса III / AIMD будут необходимыми данные клинического исследования. Клинические испытания в Европе должны быть предварительно одобрены европейским нотифицированным органом.
Назначить Европейского Авторизованного Представителя (EC REP), который расположенный в Европе.
Поместить название и адрес ЕС REP в инструкции по применению и на упаковке.
Авторизованный
Представителт
Для всех устройств, кроме Класса I (нестерильные, без функции измерения),
Ваша СМК и Технический Файл или Дизайн Досье должны пройти проверку Нотифицированным Органом, который является третьей стороной, аккредитованной Европейскими властями для оценки медицинских изделий и компаний — производящих продукты медицинского назначения.
Подготовка Декларации Соответствия, которая является документом имеющим обязательную юридическую силу, подготовленным изготовителем с указанием,
что устройство соотвечает приенимым Директивам.
Для всех медицинских изделий и средств IVD в Европейском Союзе, сертификат CE является необходим. Эта сертификация подтверждает, что устройство соответствует всем нормативным требованиям «Медицинских Директив».
CE-маркировка — регуляторные процессы Европейского Союза для медицинских изделий
*Продукция классов IIb и III с большой вероятностью потребует большого количества клинических данных. В ряде случаев могут использоваться существующие научные данные, но нередко требуется проведение клинических исследований. Клинические исследования, проводимые в ЕС должны быть одобрены Европейскими регуляторными агентствами. Планы и отчеты по клиническим испытаниям помещаются в соответствующий раздел технической документации.
**Нотифицированная Организация — организация, аккредитованная Европейским Союзом на контроль над производителями продукции медицинского назначения.
***CE-Сертификат не выдается продуктам класса I (нестерильным, без функции измерения), поскольку для медицинских изделий данного класса соответствие требованиям Регламента ЕС 2017/745 заявляется производителем в порядке самодекларации.

Скачать брошюру «CE-маркировка для медицинских изделий» в PDF-формате
Как зарегистрировать медицинское изделие в ЕС. Читать статью.
MDRC помогает производителям изделий медицинского назначения достичь соответствия регуляторным требованиям Европейского Союза, зарегистрировать и вывести на европейский рынок их продукцию.
Классификация изделий медицинского назначения
В ЕС определение класса изделий медицинского назначения осуществляется на основе ряда правил, изложенных в приложении VIII Регламента ЕС 2017/745. Поэтому медицинский продукт определенного класса по классификации РФ, США, Китая или другой страны, не входящей в Европейский Союз, в ЕС может относиться к другому классу продукции. Правильное определение класса вашей продукции является критически важным, поскольку от класса будет зависеть то, каким образом будет осуществляться регистрация и вывод на рынок ваших продуктов. Мы поможем вам правильно классифицировать вашу продукцию.
Обучение: CE-маркировка и регуляторная система ЕС для медицинских изделий
Технические файлы и досье разработки
Наличие технического файла является обязательным требованием ЕС, независимо от класса и типа продукта. Досье разработки обязательны для продуктов класса III. Досье разработки отличаются от технических файлов рядом дополнительных требований — в частности необходимостью обширной клинической программы. Мы создали многочисленные технические файлы и досье разработки для целого ряда различных продуктов и готовы помочь вам в создании технической и клинической документации для вашей продукции. Кроме того, мы проводим клиническую оценку продукции, разрабатываем инструкции по применению изделий медицинского назначения и помогаем добиться соответствия ISO 14971.
ISO 13485 и система менеджмента качества (СМК)
У MDRC имеются готовые процедуры и шаблоны документов, которые соответствуют регуляторным требованиям большинства стран и которые могут быть имплементированы в любой компании. Большинство компаний предпочитают использовать стандарт ISO 13485. Мы готовы помочь вам с внедрением СМК. Если у вас уже имеется СМК, мы поможем вам добиться ее соответствия европейским требованиям.
MDR (EU 2017/745) и IVDR (EU 2017/746) требуют наличия у производителя медицинских изделий PRRC (Person Responsible for Regulatory Compliance).
Авторизованное представительство в ЕС (EC REP)
Компании, находящиеся вне Европейского Союза, должны назначить Авторизованного Представителя в ЕС (EC REP), который будет представлять их в регуляторных органах ЕС. Хотя эту роль может выполнять дистрибьютор, наличие независимого Авторизованного Представителя несет в себе большие преимущества, поскольку позволяет менять дистрибьюторов в любое удобное для вас время. Наше германское представительство готово взять на себя роль Авторизованного Представителя для вашей компании.
Аудиты на соответствие требованиям Регламента ЕС 2017/745 и ISO 13485
Производители медицинских изделий классов IIa, IIb и III, а также стерильных продуктов класса I или изделий класса I, предназначенных для измерений, должны ежегодно проходить аудит Нотифицированной Организации. Мы поможем вам подготовиться к аудиту. Помимо этого мы можем провести для ваших работников тренинги по процедурам получения CE-марки, Регламенту ЕС 2017/745, а также внутреннему аудиту.
Взаимодействие с Нотифицированной Организацией
Производители медицинских изделий классов IIa, IIb и III, а также стерильных продуктов класса I или изделий класса I, предназначенных для измерений, взаимодействуют с регуляторными оранами ЕС через Нотифицированные Организации. За поиск и выбор Нотифицированной организации, а также коммуникацию с ней отвечает сам производитель. Мы имеем большой опыт работы с Нотифицированными Организациями из разных стран ЕС и готовы помочь вам выбрать подходящую организацию и наладить взаимодействие с ней.
Оценка и квалификация дистрибьюторов
Согласно Регламенту ЕС дистрибьюторы изделий медицинского назначения в ЕС должны отвечать целому ряду регуляторных требований. Далеко не каждая компания, готовая продавать вашу продукцию, может стать вашим дистрибьютором. В настоящий момент в Европейском Союзе — 27 стран и более 20 официальных языков. Оценка и квалификация дистрибьюторов в таких условиях — непростая задача. Мы поможем вам найти, проанализировать и отобрать подходящих дистрибьюторов.

Получение UDI и регистрация в системе EUDAMED
*Продукция классов IIb и III с большой вероятностью потребует большого количества клинических данных. В ряде случаев могут использоваться существующие научные данные, но нередко требуется проведение клинических исследований. Клинические исследования, проводимые в ЕС должны быть одобрены Европейскими регуляторными агентствами. Планы и отчеты по клиническим испытаниям помещаются в соответствующий раздел технической документации.
**Нотифицированная Организация — организация, аккредитованная Европейским Союзом на контроль над производителями продукции медицинского назначения.
***CE-Сертификат не выдается продуктам класса I (нестерильным, без функции измерения), поскольку для медицинских изделий данного класса соответствие требованиям Регламента ЕС 2017/745 заявляется производителем в порядке самодекларации.
MDRC помогает производителям изделий медицинского назначения достичь соответствия регуляторным требованиям Европейского Союза, зарегистрировать и вывести на европейский рынок их продукцию.
Классификация изделий медицинского назначения
В ЕС определение класса изделий медицинского назначения осуществляется на основе ряда правил, изложенных в приложении VIII Регламента ЕС 2017/745. Поэтому медицинский продукт определенного класса по классификации РФ, США, Китая или другой страны, не входящей в Европейский Союз, в ЕС может относиться к другому классу продукции. Правильное определение класса вашей продукции является критически важным, поскольку от класса будет зависеть то, каким образом будет осуществляться регистрация и вывод на рынок ваших продуктов. Мы поможем вам правильно классифицировать вашу продукцию.
Обучение: CE-маркировка и регуляторная система ЕС для медицинских изделий
Технические файлы и досье разработки
Наличие технического файла является обязательным требованием ЕС, независимо от класса и типа продукта. Досье разработки обязательны для продуктов класса III. Досье разработки отличаются от технических файлов рядом дополнительных требований — в частности необходимостью обширной клинической программы. Мы создали многочисленные технические файлы и досье разработки для целого ряда различных продуктов и готовы помочь вам в создании технической и клинической документации для вашей продукции. Кроме того, мы проводим клиническую оценку продукции, разрабатываем инструкции по применению изделий медицинского назначения и помогаем добиться соответствия ISO 14971.
ISO 13485 и система менеджмента качества (СМК)
У MDRC имеются готовые процедуры и шаблоны документов, которые соответствуют регуляторным требованиям большинства стран и которые могут быть имплементированы в любой компании. Большинство компаний предпочитают использовать стандарт ISO 13485. Мы готовы помочь вам с внедрением СМК. Если у вас уже имеется СМК, мы поможем вам добиться ее соответствия европейским требованиям.
Авторизованное представительство в ЕС (EC REP)
Компании, находящиеся вне Европейского Союза, должны назначить Авторизованного Представителя в ЕС (EC REP), который будет представлять их в регуляторных органах ЕС. Хотя эту роль может выполнять дистрибьютор, наличие независимого Авторизованного Представителя несет в себе большие преимущества, поскольку позволяет менять дистрибьюторов в любое удобное для вас время. Наше германское представительство готово взять на себя роль Авторизованного Представителя для вашей компании.
MDR (EU 2017/745) и IVDR (EU 2017/746) требуют наличия у производителя медицинских изделий PRRC (Person Responsible for Regulatory Compliance).
Аудиты на соответствие требованиям Регламента ЕС 2017/745 и ISO 13485
Производители медицинских изделий классов IIa, IIb и III, а также стерильных продуктов класса I или изделий класса I, предназначенных для измерений, должны ежегодно проходить аудит Нотифицированной Организации. Мы поможем вам подготовиться к аудиту. Помимо этого мы можем провести для ваших работников тренинги по процедурам получения CE-марки, Регламенту ЕС 2017/745, а также внутреннему аудиту.
Взаимодействие с Нотифицированной Организацией
Производители медицинских изделий классов IIa, IIb и III, а также стерильных продуктов класса I или изделий класса I, предназначенных для измерений, взаимодействуют с регуляторными оранами ЕС через Нотифицированные Организации. За поиск и выбор Нотифицированной организации, а также коммуникацию с ней отвечает сам производитель. Мы имеем большой опыт работы с Нотифицированными Организациями из разных стран ЕС и готовы помочь вам выбрать подходящую организацию и наладить взаимодействие с ней.
Оценка и квалификация дистрибьюторов
Согласно Регламенту ЕС дистрибьюторы изделий медицинского назначения в ЕС должны отвечать целому ряду регуляторных требований. Далеко не каждая компания, готовая продавать вашу продукцию, может стать вашим дистрибьютором. В настоящий момент в Европейском Союзе — 27 стран и более 20 официальных языков. Оценка и квалификация дистрибьюторов в таких условиях — непростая задача. Мы поможем вам найти, проанализировать и отобрать подходящих дистрибьюторов.
Получение UDI и регистрация в системе EUDAMED
Скачать брошюру «CE-маркировка для медицинских изделий» в PDF-формате
Как зарегистрировать медицинское изделие в ЕС. Читать статью.